罕见病药的价格是谁定的?10种年用药花费最高的罕见病
杭州网 发布时间:2021-09-23 09:25
国产罕见病药什么时候能研发出来?
浙大二院神经内科吴志英教授说,相较发达国家,中国的罕见病研究起步很晚。如果说罕见病的诊断与欧美国家无异,那么在治疗上,中国则远远落后于欧美国家。一方面基于很现实的原因,罕见病药物在欧美国家大部分进入医保,而国内各地的情况不尽相同。另一方面,绝大多数罕见病仍没有治疗药物,目前我们用的药几乎都来自国外大药企的发明,我们应该从源头上开始自己的创新研究,争取在治疗上有所突破。
浙大二院神经病学研究中心也在积极参与药物的研发与临床试验,但相比其他疾病,罕见病的研究经费申请非常困难。她呼吁我国能加大投入用于研究遗传病和罕见病,也希望社会各界能积极捐助罕见病的研究,寻找治疗靶点,找到更多罕见病的治疗药物,“只有这样做,我们才能和欧美比肩。”
10种年用药花费最高的罕见病排行
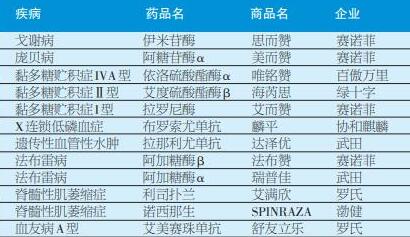
目前,按照用药年费用(包含赠药)来看,费用最高的10种罕见病为——
戈谢病-伊米苷酶
庞贝病-阿糖苷酶α
黏多糖贮积症IVA型-依洛硫酸酯酶α
黏多糖贮积症Ⅱ型-艾度硫酸酯酶β
黏多糖贮积症I型-拉罗尼酶
X连锁低磷血症-布罗索尤单抗
遗传性血管性水肿-拉那利尤单抗
法布雷病-阿加糖酶β
法布雷病-阿加糖酶α
脊髓性肌萎缩症-利司扑兰
脊髓性肌萎缩症-诺西那生
血友病A型-艾美赛珠单抗

戈谢病
戈谢病(Gaucher disease)即葡糖脑苷脂病,是一种糖脂代谢疾病,为常染色体隐性遗传,是溶酶体沉积病中最常见的一种。葡糖脑苷脂是一种可溶性的糖脂类物质,是细胞的组成成分之一,在人体内广泛存在。由于葡糖脑苷脂酶的缺乏而引起葡糖脑苷脂在肝、脾、骨骼和中枢神经系统的单核-巨噬细胞内蓄积而发病,产生相应的临床表现。
黏多糖贮积症
黏多糖贮积症是一种发病率仅2.5万分之一的罕见病。患者体内缺乏黏多糖降解酶,导致黏多糖不能被分解,积聚在体内造成各种危害。这种病又分为多种亚型,临床特点是生长迟缓、步态异常和骨骼畸形,最终将威胁生命。
X连锁低磷血症
X连锁低磷血症是一种严重的罕见遗传病,大多数患儿会经历腿部弯曲、身材矮小、骨痛和严重的牙痛。有些患者还会出现持续不适或并发症,如关节疼痛、活动能力受损、牙脓肿和听力下降。
法布雷病
法布雷病是一种十分罕见的X染色体连锁遗传的鞘糖脂类代谢疾病。发病机制是由于患者体内α-半乳糖苷酶A(α-GalA)先天性缺乏所致,从而使人体代谢产物酰基鞘氨醇三己糖苷(Gb3)和酰基鞘氨醇二己糖苷不能被裂解,在患者血管和各器官广泛蓄积,造成四肢非常剧烈的疼痛,并对肾、心脏、脑、神经等各器官产生严重损害造成病变,病情呈进行性加重发展态势,如得不到有效治疗将危及生命。
来源:都市快报 作者:记者 金晶 俞茜茜 张静 通讯员 方序 鲁青 王雪飞 编辑:高婷婷
返回